最近流行りの機械学習MD(機械学習分子動力学法)について素人が雑に説明します。
MD計算の基本
ユニットセルに入っている多数の原子・分子の動きの時間経過をシミュレーションするのがMD計算です。ではその原子をどうやって動かすかというと、原子に働く力を計算することで動かしています。力から加速度が計算され、加速度と初速度から速度が計算され、速度と現在位置から次の瞬間どこに存在しているかが計算されます。
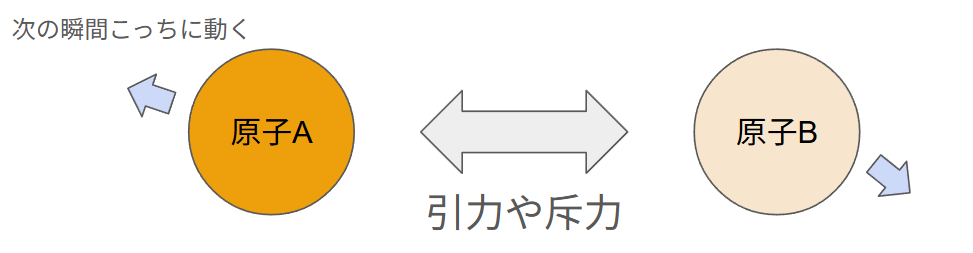
このような力や速度の計算(運動方程式)を全原子について計算して、ちょっとずつ動画のコマ送りみたいにシミュレーション内時間が進んでいきます。最終的にコマ送りをつなぎ合わせて動画のようになり、それを解析することで物質の様々な性質を知ることができます。
力をどうやって計算するか
原子に働く力と言うとファンデルワールス力やクーロン力、共有結合や金属結合など、様々なものがあり、概念としては原子の位置(配置)によってその原子に働く力が決まります。この力はポテンシャルに基づいて計算されますが、このポテンシャルとはポテンシャルエネルギー、すなわち位置エネルギーみたいな意味です。原子の位置・配置で力を計算するので力場と言ったりもします。
原子に働く力を計算する正確さ、つまりポテンシャルの精度がMD計算の精度に非常に大きく影響します。古典MD、第一原理MD、機械学習MDの3種類について比較すると以下のようになります。
| 手法 | 力の計算方法 | 特徴 |
|---|---|---|
| 古典MD | シンプルな数式と経験的なパラメータで表されるような単純な距離の関数で計算する。 原子同士の結合はバネみたいなもので代用する。 | 精度は並 大規模・長時間 化学反応は不可 |
| 第一原理MD | 経験的パラメータを使用せず、量子力学に基づいた電子状態計算から、力を高精度に計算する。 | 高精度 小規模・短時間 化学反応が再現できる |
| 機械学習MD | 第一原理から原子配置と力のパターンを学習し、力を高精度に推論する。 | 高精度 第一原理MD以上の規模・時間 化学反応が再現できる |
書き方に不正確な部分があるかもしれませんが、だいたいこんな感じです。
経験的パラメータというキーワードが出てきましたが、これは実験結果と計算結果が合うように決めたパラメータみたいな意味です。量子力学などに基づいて理論的に決めたものではなく、極論「なんかこの数字や関数を使うと結果がいい感じなんだよ」くらいのものだったりするかもしれません。
古典MDについて少しフォローすると、反応を扱えないことや力の計算精度が劣ることで化学的な現象の再現が苦手ではありますが、計算量が少なく計算速度が非常に速いため大規模・長時間の現象を扱うことができることや非力なPCでもそこそこ動くというメリットがあります。また1950年代から使われてきている歴史ある手法で、優れたポテンシャル関数やパラメータが生み出されています。目的によっては古典MDでも十分な結果が得られます。古典的な手法ですが、まだまだ現役です。
第一原理MDは高精度なMD計算ですが、計算負荷が非常に大きいです。電子状態計算には多くのCPUパワーやRAMを使用するため、大きいサンプルでは全く計算が進まないどころか、メモリ不足でPCが完全に固まってしまうこともあります。

このように、古典MDと第一原理MDは精度と計算量のトレードオフの関係があり、実際は計算対象や目的によって使い分ける必要があります。あるいは、本当は第一原理MDをやりたいけど古典MDで妥協するようなこともあるかもしれません。長時間・大規模・反応ありの計算をしたい場合、古典MDでは反応が扱えないし、第一原理MDは計算に何か月もかかってしまう…。そこで登場するのが機械学習MDです。
機械学習MDとは
機械学習MDは力の計算に機械学習(ニューラルネットワーク)を使用します。そのポテンシャルの分類からニューラルネットワークポテンシャル(NNP)、機械学習ポテンシャル(MLP)と呼ばれたりします。
計算量が非常に大きいDFT計算をニューラルネットワークによる推論で置き換えることで、第一原理MDのようなDFTレベルの精度で、古典MDのように大規模・長時間の計算ができるというのが最大のメリットです。古典MDでは再現が難しかった物質や現象を計算できるようになる、あるいは第一原理MDでは計算が難しかった規模や長時間現象の計算ができるようになります。
有名なツールとしては、Matlantis(マトランティス)、DeePMD-kit(ディープエムディーキット)、MatterSim、VASP On-the-flyなどがあります。



この3つを同列に書くのは違和感があるかもしれませんが、簡単にまとめると、
| Matlantis | 有償。 汎用ポテンシャル。 汎用:広範囲な物質を扱える。 学習済み。 GNN(グラフニューラルネットワーク)、計算は遅い。 |
| DeePMD-kit | オープンソース。 特化型ポテンシャル作成などのツール群。 特化型:対象の物質に特化したポテンシャルを作る。 ユーザーが訓練や機械学習を行う。 非GNN(記述子を使う)、計算は速い。 |
| MatterSim | オープンソース。 汎用ポテンシャル。 汎用:広範囲な物質を扱える。 学習済み。 GNN(グラフニューラルネットワーク)、計算は遅い。 |
| VASP (On-the-fly) | VASP自体は有償のソフト。その中にOn-the-fly機能がある。 特化型:第一原理MD計算を進めながら、それ自体を機械学習していく。 第一原理MDと機械学習MDを自動で賢く切り替えながら計算を進める。 |
よく知りませんがMatlantisやVASPはとても高価なので、MDや機械学習がチョットデキル方はMatterSimやDeePMD-kitを使ってみましょう。DeePMD-kitの使い方は当サイトでも何度か解説しています。


どんなときに機械学習MDを使うかですが、精度が良い古典のポテンシャルが用意できるなら古典MDで十分で、適したポテンシャルがない場合に第一原理MDや機械学習MDを検討する、という順番になると思います。
機械学習MDは万能なのか?
これまで機械学習MDの良い点ばかり紹介してきましたが、実際はそんなに簡単ではありません。例えばDeePMD-kitで機械学習MDを進める際は以下の手順になります。
- 計算対象の構造を用意する。
- 用意した構造を元に訓練データ用の構造を多数用意する。
- 訓練データ用の構造をDFT計算して訓練データを用意する。
- 機械学習をする。
- 機械学習MDを実行する。精度が悪い場合は追加訓練データを用意して3, 4, 5の手順を繰り返す。
このように手間がかかります。3.DFT計算は非常に重い計算を多数実行する必要があり、長い計算時間がかかります。これらの工程をワークフロー化してアクティブラーニングできていればいいですが、手作業でやるとなると結構面倒です。また時間をかけてNNPを作っても、MD計算の精度が悪かったり、そもそも使い物にならなかったりします。
第一原理MDの場合はどうでしょうか。
- 計算対象の構造を用意する。
- 第一原理MD計算を実行する。
手順としてはシンプルです。2.第一原理MDは非常に時間がかかりますが、逆に言うと時間をかければいいだけです。
機械学習MDと第一原理MDのそれぞれにかかる計算時間を考えた時に、
機械学習MD:|----訓練データ用のDFT計算----|-機械学習-|--機械学習MD--|
第一原理MD:|----------------------------第一原理MD----------------------------|
この2つの手法のトータルの計算時間を考えた時に、最初から力業で第一原理MDをやっていたほうが早く終わるのであれば、機械学習MDをやる意味がありません。
なお、訓練データ用のDFT計算は必ずしもMD用の計算サンプルと同じ大きさである必要はありません。数十原子程度の小規模なサンプルでDFT計算をして機械学習を行い、数百数千原子程度の大規模なサンプルで機械学習MD計算を行う、ということも可能です。もちろん小規模サンプルが十分な精度の訓練データになるというのが前提です。このような機械学習MDのメリットを生かせるのであれば機械学習MDは十分有用だと考えられます。
どういう時に第一原理MDではなく機械学習MDを使うべきか、考えてみます。
- 第一原理MDの限界を超える規模・時間の計算をする場合。
- 機械学習MDにかかるコストが第一原理MDよりも低い場合。
- 作成したニューラルネットワークポテンシャルを別の計算(似た組成の物質や、異なる温度圧力条件など)に転用できる場合。
これらの条件に合う場合は、機械学習MDを試すべきだと思います。
とは言ってもかなり手間がかかる作業になることには変わりありません。実際にやってみると、手間をかけてもゴミみたいなニューラルネットワークポテンシャルができたりします。自分で一から作りよりも、事前学習済みモデルやMatlantisを使うほうがいいかもしれません。
MatterSimはオープンソースの汎用NNPです。LAMMPSは使えないようですが、PythonのASEをよく使う人なら手軽に試せると思います。
まとめ
古典MD、第一原理MD、機械学習MDの3つの違いを大雑把に解説しました。違いは原子間に働く力の計算方法です。

質の良いニューラルネットワークポテンシャルを用意できれば、大規模・長時間の高精度なMD計算が可能になります。
コメント