PythonのASE (Atomic Simulation Environment) で赤外スペクトルを計算してみます。
計算方法
計算にはMOPACを使用します。
ASEでMOPACを使うとエラーが出る場合がありますので、その場合は以下の記事の方法で対処してください。
MOPACのハミルトニアンはPM7を使用します。(ASEのデフォルト)
MOPACで構造最適化を行い、得られた構造で赤外スペクトルを計算します。
from ase.calculators.mopac import MOPAC
from ase.optimize import BFGS
from ase.vibrations import Infrared
from ase.build import molecule
import matplotlib.pyplot as plt
#水分子の生成
atoms = molecule("H2O")
print(atoms)
print(atoms.get_all_distances())
#構造最適化
atoms.calc = MOPAC(task="1SCF GRADIENTS DISP")
optimized_geometry = BFGS(atoms)
optimized_geometry.run(fmax=0.01)
print(atoms.get_all_distances())
#赤外スペクトル計算
ir = Infrared(atoms)
ir.run()
ir.summary()
#プロット
wavenumber, intensity = ir.get_spectrum()
plt.plot(wavenumber, intensity)
plt.xlabel("Wavenumber [cm-1]")
plt.ylabel("Intensity")
plt.xlim([wavenumber[-1], wavenumber[0]]) #横軸を反転
plt.show()
これを実行します。
結果
計算は数秒で終わります。実行結果はこのようになりました。
Atoms(symbols='OH2', pbc=False)
[[0. 0.96856502 0.96856502]
[0.96856502 0. 1.526478 ]
[0.96856502 1.526478 0. ]]
Step Time Energy fmax
BFGS: 0 21:13:18 -322.674720 0.4312
BFGS: 1 21:13:18 -322.678530 0.1709
BFGS: 2 21:13:18 -322.679160 0.0236
BFGS: 3 21:13:18 -322.679180 0.0205
BFGS: 4 21:13:18 -322.679210 0.0029
[[0. 0.95521081 0.95519816]
[0.95521081 0. 1.51941545]
[0.95519816 1.51941545 0. ]]
-------------------------------------
Mode Frequency Intensity
# meV cm^-1 (D/Å)^2 amu^-1
-------------------------------------
0 4.4i 35.8i 2.4535
1 2.9i 23.1i 1.0655
2 0.1i 0.5i 0.0028
3 0.0i 0.1i 0.0001
4 0.0 0.3 0.0006
5 1.9 15.4 6.2160
6 172.9 1394.6 1.0282
7 348.4 2810.1 5.8688
8 354.1 2856.4 0.6664
-------------------------------------
Zero-point energy: 0.439 eV
Static dipole moment: 2.129 D
Maximum force on atom in `equilibrium`: 0.0029 eV/Å構造最適化の前後で.get_all_distances()をやっていますが、これで原子間距離が変化したことが確認でき、すなわちatomsオブジェクトの座標が変化したことが確認できます。
安定構造になったatomsオブジェクトでInfrared(atoms)を実行し、ir.summary()によって結果が標準出力に表示されます。
いくつか虚振動が出ていますが、重要なのは1394.6 cm-1, 2810.1 cm-1, 2856.4 cm-1です。
今回計算した水では、それぞれ変角振動、対称伸縮振動、非対称伸縮振動です。
実験で得られる赤外スペクトルは3000-3500cm-1くらいのブロードなピークが得られるものですが、計算精度としてはこんなものです。計算上は実験のようなバルク水ではなく孤立した1分子ですし。
また、結果をMatplotlibでプロットするとこうなります。
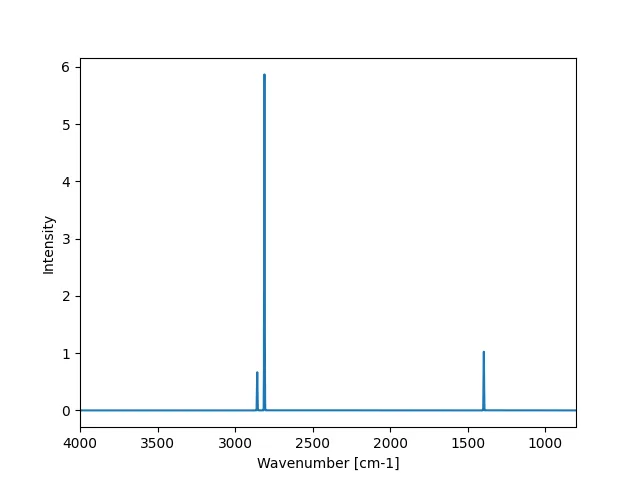
縦軸はIntensityですが、意味的には吸光度が対応するのでしょうか。
横軸は波数で、軸の左右を反転しています。
ASEでは.get_spectrum()で波数と強度の2つのリストを取得でき、デフォルトでは800-4000 cm-1の範囲で出力します。
また、計算を実行すると作業フォルダ内に赤外スペクトルの計算データフォルダが作成されます。コードを書き換えて再計算する場合はそのフォルダを消してから再計算したほうがいい場合があります。
まとめ
ASEとMOPACで水の赤外スペクトルを計算しました。
短いコードで非常に高速に計算ができました。
コメント