河村雄行先生が開発した国産MDプログラム MXDORTO(エムエックスディー・オルト で合ってるかな…)は鉱物や酸化物のMD計算で使われます。なかなか情報が少ないので、今回は使い始める際に必要な情報、ダウンロード・コンパイル・動作確認について解説していきます。
ダウンロード
NIMSのサイトからダウンロードします。

最低限必要なものは MXDORTO.zip、MXDINPUT.zipの2つです。
MXDORTOはMDの計算プログラム本体です。Serial版とMPI版があります。1行のコンパイルコマンドでビルドが完了します。ビルドに数十分かかるようなMDプログラムもある中、MXDORTOは10秒くらいでコンパイルが終わります。
MXDINPUTは入力ファイル作成ツールです。xtaldata.datもセットで使用します。結晶構造の情報や原子数、ユニットセルサイズを指定すると、それを計算するための入力ファイルを作成してくれます。コマンドラインで対話形式で動かします。Fortran特有のスペース埋め入力は、このプログラム以外では使用したことがありません。
コンパイル
ダウンロードしたzipファイルにはビルド用シェルスクリプトが入っているので、基本的にはそのシェルスクリプトを動かすだけです。
MXDORTO本体のコンパイル
Intel OneAPIのmpiifxでコンパイルを行います。
module load compiler
module load mpi
mpiifx -O2 mxdorto_p.f90 ewaldp1_new.f90 ewaldp2_new.f90 ewaldp3_new.f90 utility.f90 -lm -o bin/mxdorto_pSerial版はifxで、シェルスクリプト通りの方法でコンパイルできます。
MXDINPUTのコンパイル
mxdinputはgfortranでコンパイルします。
gfortran -O0 -Wall -g -frecursive mxdinput.f90 -o bin/mxdinputまた、xtaldata.datファイルをホームディレクトリ内の指定された場所に置く必要があります。
mkdir ~/md
mv xtaldata.dat ~/md/パスを通す
コンパイルしたmxdortoやmxdinputがあるbinディレクトリにパスを通します。実行ファイルを別のディレクトリでコンパイルした場合は、その実行ファイルを移動させてまとめておくといいと思います。
HOMEの下にmdという名前のディレクトリを作り、そこにxtaldata.dat、実行ファイルがあるとします。
必要なファイルが$HOME/mdにある場合、.bashrcの末尾に以下のものを書きます。
export PATH=~/md:$PATHcdでHOMEに戻り、source .bashrcをしたら準備完了です。
入力ファイル生成
MXDORTOは1回の計算につき1つのフォルダ(ディレクトリ)を使用します。なのでまずテスト計算用のフォルダを作ります。
$ mkdir test
$ cd test次に、ここでmxdinputを実行します。今回はアルゴンのMD計算データを作ってみます。赤字で示した部分がキーボード入力が必要な部分です。
$ mxdinput
Structure type (A12)? (see XTALDATA.DAT, or type "LIST")
............ "CHAOS" for liquid or glass
CHAOS
KEY IN AN INTEGER FOR RANDOMIZE
1
I----------------------------------------------------------I TITLE ?
test
ION1 ION2 ION3 ION4 ION5 ION6 ION7 ION8 ION9 ION0 : ATOM SPECIES ?
Ar
....1....2....3....4....5....6....7....8....9....0 NO. OF IONS ?
500
1 Ar 500 1 500
2 0 501 500
3 0 501 500
4 0 501 500
5 0 501 500
6 0 501 500
7 0 501 500
8 0 501 500
9 0 501 500
10 0 501 500
I---------I---------I---------I BOX EDGE LENGTHS (1-3) ?
50.0 50.0 50.0
-----> The density is 0.2654g/cm3. OK ? (Y/N)
y
Molecule: 1:monoatomic, 2:diatomic, or 3:H2O/CO2 (1/2/3) ?
1
Structure: 1:bulk, 2:sheet, 3:sq.rod, 4:cube, 5:sphere ?
1
Atoms: Ar
100 200 300 400 500
Random structure Ar
I--------I Temperature(K) ?
298.15
*** NO. of ions : Ar: 500 : 0 : 0 : 0 : 0
: 0 : 0 : 0 : 0 : 0
*** A= 50.0000 B= 50.0000 C= 50.0000 CA= 0.0000 CB= 0.0000 CC= 0.0000
*** Density is 0.2654 g/cm3
Do you want a new file05.dat (y/n/r) ?
yRANDOMIZEに入れる数字は乱数種です。適当な数字を入れましょう。
また入力する際は文字列の幅を意識する必要があり、幅に合わせてスペース埋めをします。
I---------I
このIとIの間に数字や文字を入力する。
ION1 ION2 ION3
ここもIONのIの位置に合わせて元素名を入力する。
....1....2....3
ここも同様に各元素の数を、位置を合わせて入力する。このようにして、50Å×50Å×50Åのボックスの中にArが500個入り、298.15Kの温度条件で計算をするファイルを作成しました。testフォルダ内にfile05.datとfile07.datが作成されます。入力ファイルはこの2つです。
| file05.dat | 計算条件とポテンシャル設定 |
| file07.dat | ユニットセルサイズと原子座標 |
mxdinputによってこの2つのファイルが作られます。この2つはテキストファイルなので、これらのファイルを書き換えることで計算設定や構造を変えることができます。
file05.dat
file05.datの内容はこのようになっています。
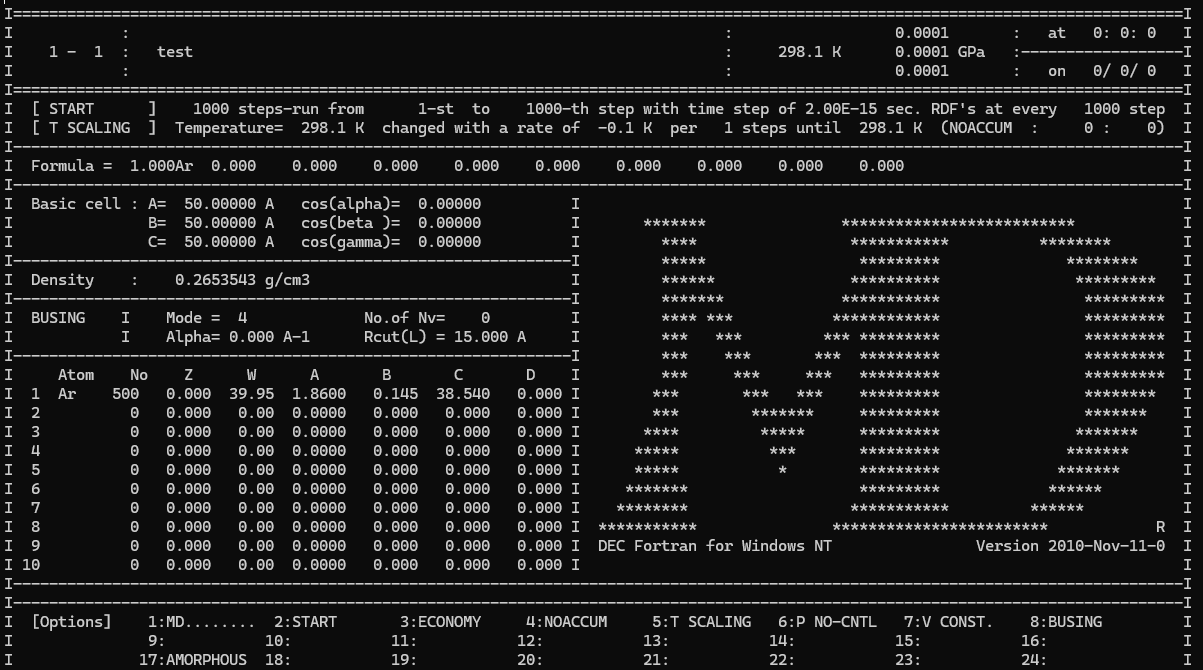
MD.......I....:....I....:....I....:....I....:....I....:....I....:....I:
START test :
ECONOMY 01000. 1000. 100. 50. 5. :
NOACCUM 2.00 1.0 0.0 :
T SCALING 298.15 -0.1 1. :
P NO-CNTL 0.0001 0.0001 0.0001 :
V CONST. :
BUSING 4. 0.0 :
1 Ar 500. 0.000 39.95 1.860 0.145 38.540 :
:
:
MD.......I....:....I....:....I....:....I....:....I....:....I....:....I:
STOPとりあえずこのまま動かしましょう。入力ファイルの書式はそのうち別記事で解説します。
file07.dat
file07.datはこのような内容です。こちらの書き方も別記事で解説します。
test 0 0
500 1 0 0 0 0 0 0 0 0 0
Ar
500 0 0 0 0 0 0 0 0 0
1 501 501 501 501 501 501 501 501 501
500 500 500 500 500 500 500 500 500 500
298.15 -1.0000 298.15 0.00010 0.00010 0.00010
0.20E-14 50.000000 50.000000 50.000000 0.000000 0.000000 0.000000
0.265354 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.818959960.393459970.77533997 4.97951175.01372394.9836850 0.8189600 0.3934600 0.7753400 1 0
0.068320000.594020000.27239000 4.99428274.97679955.0241919 0.0683200 0.5940200 0.2723900 1 0計算実行
では計算を実行します。1つの計算につき1つのフォルダが対応します。フォルダ内にfile05.datとfile07.datがある状態で計算を実行します。
Serial版ならmxdorto、MPI版ならmpiexec -n 2 mxdorto_pのコマンドを実行します。
$ ls
file05.dat file07.dat
$ mxdorto
Welcome to MOLECULAR DYNAMICS SIMULATION WORLD: MD-XD-ORTHO Version 2025-11-20-
============== Titles in FILE07.DAT and FILE05.DAT are ==============
=====[F7]: test =====
=====[F5]: test =====
Option[ 1:MD...... 2:START 3:ECONOMY 4:NOACCUM 5:T SCALIN 6:P NO-CNT]
[ 7:V CONST. 8:BUSING 9: 10: 11: 12: ]
[13: 14: 15: 16: 17:AMORPHOU 18: ]
[19: 20: 21: 22: 23: 24: ]
[25: 26: 27: 28: 29: 30: ]
[31: 32: 33: 34: 35: 36: ]
[37: 38: 39: 40: 41: 42: ]
[43: 44: 45: 46: 47: 48: ]
[49: 50: 51: 52: 53: 54: ]
Started at 23:34:35 on 2026/ 1/23 from 1-st step, until 1000-th step
== test (End= 1000) ==
+ 00K steps ----------------------------------------------------------- Hour=23
STEP Temp Prss.( Px Py Pz ) U(Coul.) U(srt) U(3p) E(total) Density mn:sc
1 297 0.0142( 0.01 0.02 0.01) 0.000 -1.142 0.000 2.575 0.26535 34'35
2 297 0.0143( 0.01 0.02 0.01) 0.000 -1.141 0.000 2.576 0.26535 34'35
3 297 0.0143( 0.01 0.02 0.01) 0.000 -1.140 0.000 2.577 0.26535 34'35
4 297 0.0143( 0.01 0.02 0.01) 0.000 -1.138 0.000 2.578 0.26535 34'35
5 297 0.0144( 0.01 0.02 0.01) 0.000 -1.137 0.000 2.579 0.26535 34'35
Msd: 0.002(A) 0.000( ) 0.000( ) 0.000( ) 0.000( )
6 297 0.0144( 0.01 0.02 0.01) 0.000 -1.136 0.000 2.580 0.26535 34'35
:
:
999 298 0.0156( 0.01 0.02 0.02) 0.000 -1.092 0.000 2.628 0.26535 34'37
1000 298 0.0156( 0.01 0.02 0.02) 0.000 -1.092 0.000 2.627 0.26535 34'37
Msd: 51.027(A) 0.000( ) 0.000( ) 0.000( ) 0.000( )
--------------------------------------------------------------------------------
Avr 298 0.0138( 0.01 0.02 0.01) 0.00 -1.14 0.00 2.57 0.26535
Std 0 0.0010( 0.00 0.00 0.00) 0.00 0.03 0.00 0.03 0.00000
--------------------------------------------------------------------------------
Temperatures: Ar: 298
--------------------------------------------------------------------------------
=============== Files were updated ============ End= 1000 ===============
<<<= ==== test ====>>>
=========================================================================
===== Started at 23:34:35 on 1/23, finished at 23:34:37 on 1/23 =====
=========================================================================計算は正常に終了しました。計算中の出力には温度圧力などが表示されます。計算後にはfile06.datなどが増えます。また構造データであるfile07.datは最終スナップショットの構造で上書きされます。
$ ls
file05.dat file06.dat file07.dat file08.dat file081.dat file09p.dat file09v.dat各ファイルは以下のような内容です。
| 入力 | file05.dat | 計算条件とポテンシャル設定 |
| 出力 | file06.dat | 計算結果まとめ |
| 入力&出力 | file07.dat | ユニットセルサイズと原子座標 |
| 出力 | file08.dat | 二体相関関数 |
| 出力 | file081.dat | 温度やエネルギー等の結果 |
| 出力 | file09p.dat | 原子座標(トラジェクトリ) |
| 出力 | file09v.dat | 原子速度 |
出力ファイルの中にはリスタート計算時には入力ファイルになるものもあります。
計算結果の読みやすいものはfile06.datに記録されます。中には温度やエネルギー等の時間変化や二体相関関数が書かれています。今回は動作確認ということで意味のある計算はしていませんので、結果の解説はしません。
ダウンロード・コンパイル・動作確認が完了したということで今回はここまで。
コメント