PythonのRDKitを使って、SMILES表記の分子からXYZ座標のMOPAC入力ファイルを作ります。
環境
Python 3.11.4
RDKit 2023.9.2
MOPAC v22.1.0
RDKitをインストールしていない場合はpipか何かで入れましょう。
pip install rdkitMOPACのインストール方法はこちらです。
書いたコードと実行結果
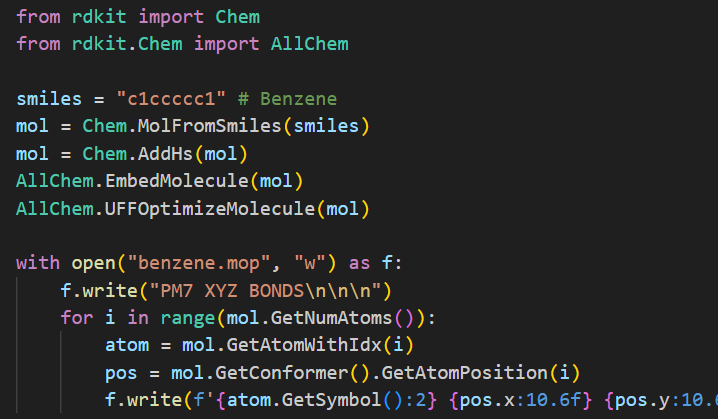
書いたコードはこんな感じです。
ベンゼン(c1ccccc1)を入力としていますが、たくさんある場合はリストに入れてforループで回すといいと思います。
from rdkit import Chem
from rdkit.Chem import AllChem
smiles = "c1ccccc1"
mol = Chem.MolFromSmiles(smiles)
mol = Chem.AddHs(mol)
AllChem.EmbedMolecule(mol)
AllChem.UFFOptimizeMolecule(mol)
with open("benzene.mop", "w") as f:
f.write("PM7 XYZ BONDS\n\n\n")
for i in range(mol.GetNumAtoms()):
atom = mol.GetAtomWithIdx(i)
pos = mol.GetConformer().GetAtomPosition(i)
f.write(f'{atom.GetSymbol():2} {pos.x:10.6f} {pos.y:10.6f} {pos.z:10.6f}\n')SMILES形式を読み込んで、水素を付加して、なんかいい感じの3次元構造にしてファイルに書き出す、という流れです。
これでbenzene.mopというファイルが作られます。
PM7 XYZ BONDS
C 0.806498 -1.143092 0.014916
C 1.393280 0.126835 -0.002138
C 0.586782 1.269928 -0.017050
C -0.806497 1.143093 -0.014912
C -1.393280 -0.126835 0.002138
C -0.586782 -1.269928 0.017055
H 1.430428 -2.027420 0.026448
H 2.471161 0.224957 -0.003790
H 1.040732 2.252381 -0.030245
H -1.430426 2.027421 -0.026450
H -2.471161 -0.224959 0.003787
H -1.040735 -2.252379 0.030241
1行目はキーワードで、この場合はPM7で構造最適化が実行されます。
このファイルをMOPACで計算します。
PATHが通っていれば以下のコマンドで動きます。
mopac benzene.mop結果を一部抜粋すると、
:
:
:
CARTESIAN COORDINATES
1 C 0.803571743 -1.138816331 0.014866065
2 C 1.388379109 0.126399553 -0.002131921
3 C 0.584645654 1.265212839 -0.016985166
4 C -0.803557543 1.138885429 -0.014858728
5 C -1.388201441 -0.126346917 0.002135744
6 C -0.584701246 -1.265323113 0.016984697
7 H 1.430564173 -2.027866184 0.026457641
8 H 2.471432749 0.225030246 -0.003791121
9 H 1.040916978 2.252752661 -0.030258085
10 H -1.430868321 2.027798415 -0.026454425
11 H -2.471595095 -0.224964453 0.003791091
12 H -1.040918468 -2.252785794 0.030240066
:
:
:
FINAL HEAT OF FORMATION = 22.95549 KCAL/MOL = 96.04578 KJ/MOL
COSMO AREA = 119.69 SQUARE ANGSTROMS
COSMO VOLUME = 108.37 CUBIC ANGSTROMS
GRADIENT NORM = 0.41904 = 0.12097 PER ATOM
IONIZATION POTENTIAL = 9.824125 EV
HOMO LUMO ENERGIES (EV) = -9.824 0.236
NO. OF FILLED LEVELS = 15
MOLECULAR WEIGHT = 78.1134 POINT GROUP: D6h
:
:
:
**********************
* *
* JOB ENDED NORMALLY *
* *
**********************
TOTAL JOB TIME: 0.19 SECONDS
== MOPAC DONE ==計算は一瞬で終わります。
結果ファイルのbenzene.outを開くといろいろ確認できます。
- 最適化された座標
- CHARGE:部分電荷
- DIPOLE:双極子
- FINAL HEAT OF FORMATION:生成熱
- COSMO VOLUME:分子体積(溶媒体積)
- BOND ORDER:結合次数
などなど。計算時に付けるキーワードによって結果の出力項目は変わります。
まとめ
RDKitを使ってSMILESからMOPAC計算ファイルを作成しました。
コメント