PythonのASE (Atomic Simulation Environment) で分子を作成する際の基本について簡単にまとめたいと思います。
<class ‘ase.atoms.Atoms’>
分子はこのクラスのオブジェクトです。試しに酢酸分子を作ってみましょう。
from ase.build import molecule
atoms = molecule("CH3COOH")
pos = atoms.get_positions()
elm = atoms.get_chemical_symbols()
num = atoms.get_atomic_numbers()
mas = atoms.get_masses()
print(atoms.symbols)
for i in range(atoms.get_global_number_of_atoms()):
print("%3i %3s %3i %8.3f"%(i, elm[i], num[i], mas[i]), pos[i])
print(mas.sum())この「atoms」が<class ‘ase.atoms.Atoms’>のオブジェクト、すなわち分子です。
このクラスで使えるメソッドはたくさんありますが、ここで使っているものは
- .get_positions() 原子数 × XYZ座標の二次元配列
- .get_chemical_symbols() 元素記号
- .get_atomic_numbers() 原子番号
- .get_masses() 原子量
出てくるものはNumpyArrayなので、例えば最後にprintしているように原子量の.get_masses()を.sum()すると分子量になります。
実行結果はこうなります。
CO2HCH3
0 C 6 12.011 [0. 0.15456 0. ]
1 O 8 15.999 [0.166384 1.360084 0. ]
2 O 8 15.999 [-1.236449 -0.415036 0. ]
3 H 1 1.008 [-1.867646 0.333582 0. ]
4 C 6 12.011 [ 1.073776 -0.892748 0. ]
5 H 1 1.008 [ 2.048189 -0.408135 0. ]
6 H 1 1.008 [ 0.968661 -1.528353 0.881747]
7 H 1 1.008 [ 0.968661 -1.528353 -0.881747]
60.05199999999999なお、from ase.build import molecule の molecule("CH3COOH") のように名前で指定できるものの一覧はこちらです。
>>> from ase.collections import g2
>>> print(g2.names)
['PH3', 'P2', 'CH3CHO', 'H2COH', 'CS', 'OCHCHO', 'C3H9C', 'CH3COF',
'CH3CH2OCH3', 'HCOOH', 'HCCl3', 'HOCl', 'H2', 'SH2', 'C2H2', 'C4H4NH',
'CH3SCH3', 'SiH2_s3B1d', 'CH3SH', 'CH3CO', 'CO', 'ClF3', 'SiH4',
'C2H6CHOH', 'CH2NHCH2', 'isobutene', 'HCO', 'bicyclobutane', 'LiF',
'Si', 'C2H6', 'CN', 'ClNO', 'S', 'SiF4', 'H3CNH2', 'methylenecyclopropane',
'CH3CH2OH', 'F', 'NaCl', 'CH3Cl', 'CH3SiH3', 'AlF3', 'C2H3', 'ClF',
'PF3', 'PH2', 'CH3CN', 'cyclobutene', 'CH3ONO', 'SiH3', 'C3H6_D3h',
'CO2', 'NO', 'trans-butane', 'H2CCHCl', 'LiH', 'NH2', 'CH',
'CH2OCH2', 'C6H6', 'CH3CONH2', 'cyclobutane', 'H2CCHCN', 'butadiene',
'C', 'H2CO', 'CH3COOH', 'HCF3', 'CH3S', 'CS2', 'SiH2_s1A1d', 'C4H4S',
'N2H4', 'OH', 'CH3OCH3', 'C5H5N', 'H2O', 'HCl', 'CH2_s1A1d', 'CH3CH2SH',
'CH3NO2', 'Cl', 'Be', 'BCl3', 'C4H4O', 'Al', 'CH3O', 'CH3OH', 'C3H7Cl',
'isobutane', 'Na', 'CCl4', 'CH3CH2O', 'H2CCHF', 'C3H7', 'CH3', 'O3',
'P', 'C2H4', 'NCCN', 'S2', 'AlCl3', 'SiCl4', 'SiO', 'C3H4_D2d',
'H', 'COF2', '2-butyne', 'C2H5', 'BF3', 'N2O', 'F2O', 'SO2', 'H2CCl2',
'CF3CN', 'HCN', 'C2H6NH', 'OCS', 'B', 'ClO', 'C3H8', 'HF',
'O2', 'SO', 'NH', 'C2F4', 'NF3', 'CH2_s3B1d', 'CH3CH2Cl',
'CH3COCl', 'NH3', 'C3H9N', 'CF4', 'C3H6_Cs', 'Si2H6', 'HCOOCH3',
'O', 'CCH', 'N', 'Si2', 'C2H6SO', 'C5H8', 'H2CF2', 'Li2', 'CH2SCH2',
'C2Cl4', 'C3H4_C3v', 'CH3COCH3', 'F2', 'CH4', 'SH', 'H2CCO',
'CH3CH2NH2', 'Li', 'N2', 'Cl2', 'H2O2', 'Na2', 'BeH', 'C3H4_C2v', 'NO2']化合物名やSMILESやCIDで分子を作成
PubChemを使って分子を作ることもできます。
from ase.data.pubchem import pubchem_atoms_search
benzene1 = pubchem_atoms_search(name="benzene")
benzene2 = pubchem_atoms_search(cid=241)
benzene3 = pubchem_atoms_search(smiles="c1ccccc1")
print(benzene1.symbols)
print(benzene1 == benzene2, benzene1 == benzene3)結果はこうなります。
C6H6
True True化合物名、CID、SMILES、いずれも同じオブジェクトが作られました。
また、複数の配座がある場合は、
from ase.data.pubchem import pubchem_atoms_search, pubchem_atoms_conformer_search
octane_conformers = pubchem_atoms_conformer_search(name="octane")このoctane_conformersはPythonのリストなので、octane_conformers[0]のようにして1個目の分子のオブジェクトを選択できます。
座標を入力して作る場合
二酸化炭素の分子を作ってみます。
from ase import Atoms
CO2 = Atoms("OCO", positions=[(-1.16, 0, 0), (0, 0, 0), (1.16, 0, 0)])
print(CO2.get_chemical_symbols())
print(CO2.get_masses().sum())
print(CO2.get_all_distances())結果はこうなります。
['O', 'C', 'O']
44.009
[[0. 1.16 2.32]
[1.16 0. 1.16]
[2.32 1.16 0. ]]最後の.get_all_distances()は各原子同士の距離を出力します。
2番目の[1.16 0. 1.16]を見ると、Cの原子が2つのOに対して1.16Åの距離にあることが分かります。
可視化
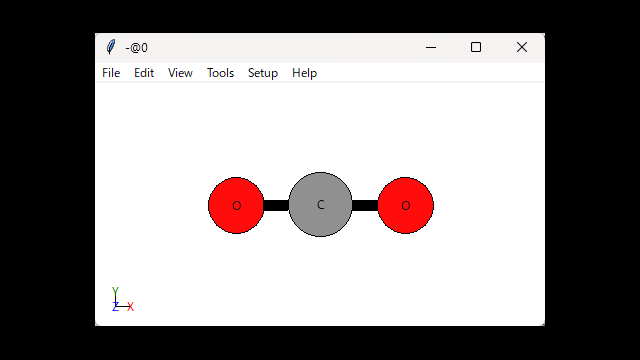
先ほどのCO2分子を可視化します。
from ase import Atoms
from ase.visualize import view
CO2 = Atoms("OCO", positions=[(-1.16, 0, 0), (0, 0, 0), (1.16, 0, 0)])
view(CO2)実行するとViewerが出てきます。
View設定をするとこんな感じ。
(Bond情報ってどこにあるんだろう…)
ファイルのインポート
GAMESS_USの計算結果をインポートしてみます。
from ase.io import read
atoms = read("benzene.out")
print(atoms.get_atom_positions())結果はこうなります。
[[ 0.64489995 -1.23569991 0. ]
[ 1.3927999 -0.0594 0. ]
[ 0.74779995 1.17629991 0. ]
[-0.64489995 1.23579991 0. ]
[-1.3926999 0.0595 0. ]
[-0.74789995 -1.17639991 0. ]
[ 1.14799992 -2.20039984 0. ]
[ 2.47939982 -0.10579999 0. ]
[ 1.3313999 2.09439985 0. ]
[-1.14829992 2.20029984 0. ]
[-2.47959982 0.10589999 0. ]
[-1.3314999 -2.09449985 0. ]]このoutファイルはGAMESSの構造最適化の結果なのですが、実は最適化された座標ではなく初期座標が読み込まれています。
構造最適化の結果の座標を読み込むことはできないようです。
GAMESSで構造最適化をしたい場合はASE上で構造最適化を実行する必要があります。
詳しくはこちらの記事で解説しています。
コメント