PythonのASE (Atomic Simulation Environment) で分子を作成してGAMESSで構造最適化する方法について簡単にまとめたいと思います。
準備
いくつか必要な作業があります。
GAMESSインストール
すでにインストール済みなら不要です。Windows版のインストール方法はこちら。
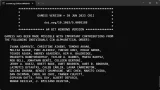
環境変数
環境変数 ASE_GAMESSUS_COMMAND を作成し、C:\Users\Public\gamess-64\rungms.bat PREFIX.inp 2023.R1.intel 1 > PREFIX.log 2> PREFIX.err
のような形で設定します。
順序としては[rungms.batのパス] [input] [バージョン名] [並列数]… みたいな感じです。
rungms.gmsのコピー
これはもっといい方法がある気がしますが、とりあえずこうしています。
C:\Users\Public\gamess-64\rungms.gms のファイルを、Pythonを実行するカレントディレクトリにコピーします。
これをせずにPythonのASEを実行するとGAMESSのエラーが出ます。
コード
二酸化炭素の分子の構造最適化をやってみます。コードは以下のようになりました。
from ase.build import molecule
from ase.calculators.gamess_us import GAMESSUS
from ase.optimize import BFGS
atoms = molecule("CO2")
print(atoms.get_chemical_symbols())
print(atoms.get_all_distances())
atoms.calc = GAMESSUS(userscr="C:\\Users\\Public\\gamess-64\\scratch")
optimized_geometry = BFGS(atoms)
optimized_geometry.run()
print(atoms.get_all_distances())
こんな感じで、ASEが作成したCO2分子の原子間距離とGAMESSで構造最適化したCO2分子の原子間距離を比較します。
分子の作り方はこちらで解説しています。
GAMESSUS(userscr="C:\\Users\\Public\\gamess-64\\scratch")の部分でUSRSCRディレクトリの場所を指定しています。userscrは書かなくても動きますが警告っぽいものが出ます。
また指定する場合は区切りが\マークが2個になっているのでご注意ください。
最適化自体はGAMESSではなく10行目にあるBFGSが実行していて、原子間に働く最大のForce(fmax)が一定以下になると収束判定されます。この閾値は11行目のrunが持っていて、デフォルトでは.run(fmax=0.05)が指定されています。この単位は[eV/A]です。
実行結果はこうなりました。
['C', 'O', 'O']
[[0. 1.178658 1.178658]
[1.178658 0. 2.357316]
[1.178658 2.357316 0. ]]
Step Time Energy fmax
BFGS: 0 22:19:02 -5076.530826 2.5319
BFGS: 1 22:19:08 -5076.568619 1.6673
BFGS: 2 22:19:14 -5076.590259 0.1301
BFGS: 3 22:19:19 -5076.590401 0.0060
[[0. 1.15580988 1.15580988]
[1.15580988 0. 2.31161977]
[1.15580988 2.31161977 0. ]]最初と最後にある.get_all_distances()では3つの原子同士のそれぞれの距離を出力しています。
2行目にある3つが[C-C, C-O, C-O]の距離です。C自身は0.0で、Cと各Oとの距離が1.178658Åです。
最適化のサイクルは0stepから3stepまで進んでfmaxが0.05以下になり収束しました。
最適化後のC-O距離は1.15580988Åです。
計算設定
先ほどの例ではデフォルト設定でGAMESSの計算が実行されました。
最適化サイクルの3step目のファイルが残っていたので見てみます。
$CONTRL
RUNTYP=GRADIENT
MULT=1
SCFTYP=RHF
$END
$BASIS
GBASIS=N21
NGAUSS=3
$END
$DATA
CO2
C1
C 6 0.0000000000000e+00 0.0000000000000e+00 0.0000000000000e+00
O 8 0.0000000000000e+00 0.0000000000000e+00 1.1558098828031e+00
O 8 0.0000000000000e+00 0.0000000000000e+00 -1.1558098828031e+00
$ENDGBASIS=N21, NGAUSS=3なので3-21Gですね。これがデフォルトです。
変更するには、コードのGAMESSUS(userscr="C:\\Users\\Public\\gamess-64\\scratch")の部分を以下のようにします。
atoms.calc = GAMESSUS(userscr="C:\\Users\\Public\\gamess-64\\scratch",
contrl = dict(dfttyp="B3LYP"),
basis = dict(gbasis="n31", ngauss=6))この場合は B3LYP 6-31G で計算されます。
CO2のC-O距離は1.18827217Åになりました。
まとめ
PythonのASEを使ってGAMESSで構造最適化計算ができました。
ASEでは分子モデリングも可視化もできるので、GAMESSのGUIツールと言っても過言ではないですね。
コメント