
機械学習力場のツールであるDeePMD-kitで作成したポテンシャルでMD計算を試してみます。
Part.1とPart.2の続きです。

LAMMPS用の構造ファイルの作成
作成した graph-compress.pb はダイヤモンド(C 64個)を学習させたものでしたので、とりあえず同じものを計算してみます。LAMMPSのdataファイルはASEで作ります。
from ase.build import bulk
from ase.units import mol
diamond = bulk("C", "diamond", cubic=True)
n=2
unitcell = diamond.repeat((n, n, n))
density = (np.sum(unitcell.get_masses())/mol) / (unitcell.get_volume()*1e-24)
print(density, "[g/cm3]")
unitcell.write("diamond_lammps.data", format='lammps-data')
Part.1のコードとほぼ一緒です。これでdataファイルができます。
そしてこのdataファイルと一緒に使用するinファイルは以下のものを使用します。
units metal
boundary p p p
atom_style atomic
neighbor 1.0 bin
neigh_modify every 10 delay 0 check no
read_data diamond_lammps.data
mass 1 12.011
pair_style deepmd graph-compress.pb
pair_coeff * *
velocity all create 300.0 23456789
fix 1 all npt temp 300.0 300.0 0.1 tri 1.0 1.0 1.0
timestep 0.001
thermo_style custom step pe ke etotal temp press vol density
thermo 100
dump 1 all custom 100 diamond_lammps.dump id type x y z
run 50000DeePMD-kitのポテンシャルを使用する際は単位系が units metal になります。このinファイル中では、時間の単位が ps になっています。
LAMMPSで計算
このinとdataをLAMMPSで計算します。
docker版DeePMD-kitを使っている場合はdockerの中でlmpコマンドが使用できます。
mpiexec -n 8 lmp -in diamond_lammps.indockerのGPU版DeePMD-kitを使用した場合、このコマンドでCPUとGPUが両方使用されていました。計算開始時にDeePMD関連の出力がたくさん出てきて、少し待つと計算が始まりました。
#計算の最後のほう
49900 -16043.126 2.1810228 -16040.945 267.8274 116255.85 365.86561 3.4888846
50000 -16043.115 2.0856728 -16041.029 256.11852 97895.456 366.03 3.4873177
Loop time of 825.452 on 8 procs for 50000 steps with 64 atoms
Performance: 5.233 ns/day, 4.586 hours/ns, 60.573 timesteps/s, 3.877 katom-step/s
96.4% CPU use with 8 MPI tasks x 1 OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 736.71 | 747.11 | 758.14 | 23.5 | 90.51
Neigh | 0.29642 | 0.2988 | 0.3006 | 0.3 | 0.04
Comm | 64.896 | 75.923 | 86.32 | 73.7 | 9.20
Output | 0.041678 | 0.046539 | 0.069008 | 4.0 | 0.01
Modify | 1.9141 | 1.9325 | 1.9525 | 0.8 | 0.23
Other | | 0.1427 | | | 0.02
Nlocal: 8 ave 12 max 6 min
Histogram: 1 4 0 1 0 0 1 0 0 1
Nghost: 856 ave 858 max 852 min
Histogram: 1 0 0 1 0 0 1 0 4 1
Neighs: 0 ave 0 max 0 min
Histogram: 8 0 0 0 0 0 0 0 0 0
FullNghs: 2191.75 ave 3288 max 1643 min
Histogram: 1 4 0 1 0 0 1 0 0 1
Total # of neighbors = 17534
Ave neighs/atom = 273.96875
Neighbor list builds = 5000
Dangerous builds not checked
Total wall time: 0:13:52計算時間の大部分を Pair のSectionが占めています。機械学習の結果に基づいて原子の位置関係から直接エネルギーを予測しているので、この処理の計算負荷が大きいです。今回はGPUが動いていたのでCPU onlyと比べると少しは速かったのでしょうか。
計算速度は原子が64個しかないMD計算として見ると非常に遅いですが、QEの第一原理MDと同等のものが動いているのだとしたら非常に速いです。果たして結果は同等と言えるのでしょうか?
結果
LAMMPSの結果のアニメーションはこちらです。
これが良いか悪いかは分かりませんが、少なくとも結晶構造を保っていることは確認できました。
次に密度を見てみます。
QE pw.x
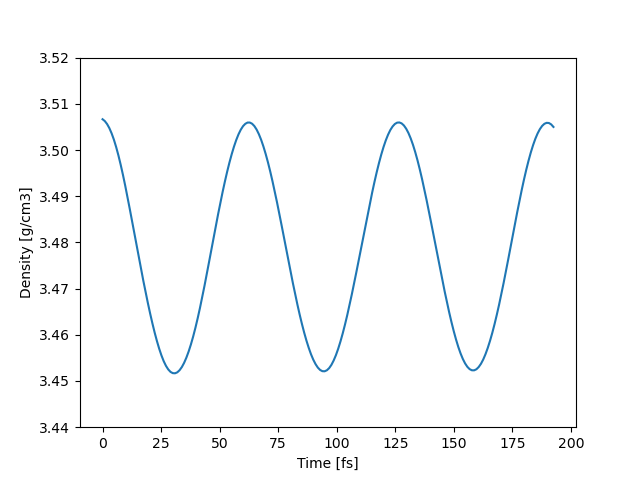
平均密度:3.4792 g/cm3
密度は cat diamond.out | grep “density =” で確認した。
LAMMPS DeePMD-kit
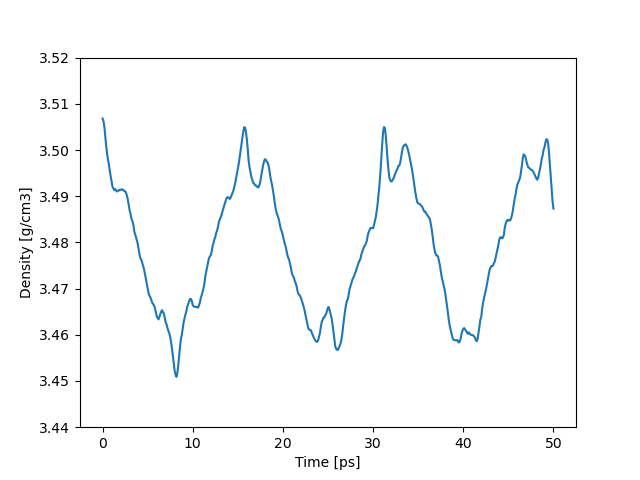
平均密度:3.4795 g/cm3
密度は log.lammps に載っている。
時間スケールがかなり違いますが、揺らぎの周期が同じに見えます。(偶然です)
縦軸のスケールは同じですが、このように同じレンジで密度が揺らいでいます。また平均値もかなり一致しています。
まとめ
Quantum ESPRESSOのpw.xによる第一原理MD計算を機械学習してポテンシャルを作成し、それを使ったLAMMPSのMD計算で同等の密度を再現することができました。pw.xによるMD計算結果はそのままDeePMD-kitに読み込ませることはできませんが、間にASEの処理を挟むことでデータを読み込ませることができました。
計算対象が非常にシンプルなものだったので結果も良かったですが、多数の元素が入ったサンプルで実用的なものを作ろうと思うと多くの時間と手間と計算資源が必要になりそうです。
Part.1から続くシリーズは今回のPart.3が最後です。今回の3記事が「とりあえずDeePMD-kitを動かしてみたい!」「QEのデータを使ってみたい!」という方の参考になれば幸いです。
コメント
Part1~Part3まで拝見し大変参考になりました。
Q1
今回、300Kのpw.xの計算で教師データを作成された内容となっていますが、例えば、複数の温度(300K、500K、800Kなど)の計算結果から教師データを作成することは出来ないでしょうか?
Q2
また、「cp.xのほうだとトラジェクトリで読んでくれます」ということですが、cp.xで教師データを作成する方法もご教授頂けますと幸いです。
以上、宜しくお願い致します。
どちらのご質問も少し検証すれば分かりそうなので、いつになるかは分かりませんがまた記事にしたいと思います。
酸化物などでやってみてるんですが、deepmdで作成した力場で密度がなかなか合いません。
一方、aenetでは、密度も弾性率も合いました。
deepmdの評判はどんな感じなのでしょうか?
aenet使ったことなかったです。評判は分かりませんが、やはり複数元素の計算は難しいんですね。
はい、何となくですけど、deepmdは複数の種類の元素があると、物性値と合わない気がします。