PythonのASE (Atomic Simulation Environment) でLAMMPSのデータファイルを作って可視化してみます。
書いたコード
メタノール(CH3OH)の分子を64個作ってみます。
from ase import Atoms
from ase.visualize import view
from ase.build import molecule
import numpy as np
methanol = molecule("CH3OH")
print(methanol.get_chemical_symbols())
unitcell_size = 5.0
unitcell = Atoms(symbols=methanol.get_chemical_symbols(), positions=methanol.get_positions(),
cell=np.eye(3) * unitcell_size, pbc=True)
mdcell = unitcell * [4, 4, 4]
mdcell.write("methanol_64.data", format='lammps-data', units="real", atom_style="full")
view(mdcell)
methanolがメタノール分子、それを1つセルの中に入れてunitcellとしています。unitcell_sizeの5.0は適当です。それを4x4x4で積層しています。4x4x4で64個の分子ができます。
積層したセルがmdcellで、これに対して.write(ファイル名, format=”lammps-data”) を実行することでLAMMPSのデータファイルが出力されます。
ここでunitsとatom_styleを指定しています。atom_styleはatom, full, chargeの中から選べます。
また最後にviewで可視化します。
実行
実行するとLAMMPSのデータが出力され、構造のViewerが出てきます。
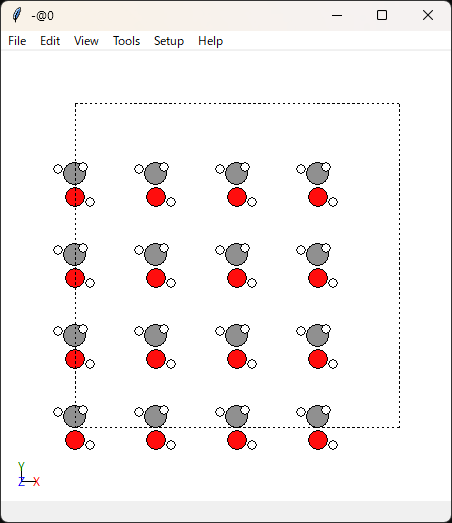
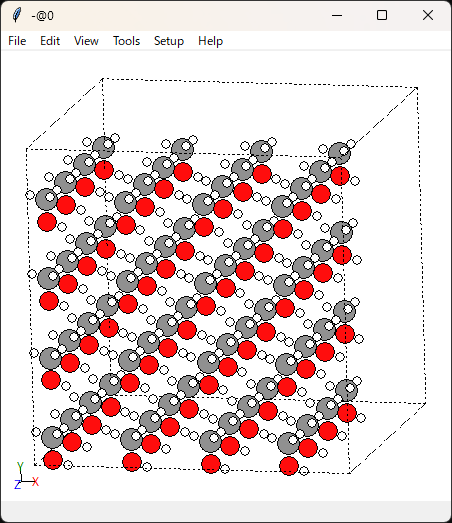
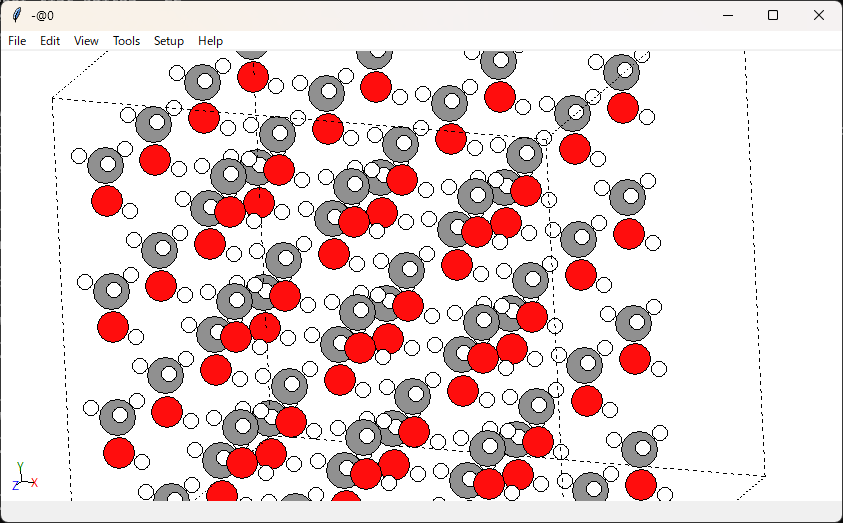
こんな感じで可視化ができます。右クリックを押しながら回転させられます。
分子同士の距離は大丈夫そうですね。あまり近すぎると計算が破綻しますし、遠すぎると密度緩和に時間がかかってしまいます。
unitcell_size=5.0で分子1個当たりのセルサイズを指定していたので、問題がある場合はこの値を調整します。
出力したLAMMPSのデータファイルは以下の通りです。
methanol_64.data (written by ASE)
384 atoms
3 atom types
0.0 20 xlo xhi
0.0 20 ylo yhi
0.0 20 zlo zhi
Atoms
1 0 1 0.0 -0.047130999999999999 0.66438900000000001 0
2 0 3 0.0 -0.047130999999999999 -0.75855099999999998 0
3 0 2 0.0 -1.0929949999999999 0.96978500000000001 0
4 0 2 0.0 0.87853400000000004 -1.0484579999999999 0
5 0 2 0.0 0.43714500000000001 1.080376 0.89177200000000001
6 0 2 0.0 0.43714500000000001 1.080376 -0.89177200000000001
7 0 1 0.0 -0.047130999999999999 0.66438900000000001 5
8 0 3 0.0 -0.047130999999999999 -0.75855099999999998 5
以下略このように原子の座標が書かれます。
Atomsセクションは左から順に原子ID、分子ID、元素、部分電荷、X Y Z座標です。
元素が数字で書かれていて分からないので解読します。
先ほど実行したプログラムではprint文で [‘C’, ‘O’, ‘H’, ‘H’, ‘H’, ‘H’] を出力していましたが、このAtomsはこの順番に書かれています。
つまりこうなります。
| C | O | H | H | H | H |
| 1 | 3 | 2 | 2 | 2 | 2 |
というわけで、元素を1 2 3の順で書くとC H Oとなります。
ちなみにこのデータファイルにはBondはなく、分子グループの定義もされていません。
ファイル出力をfullで行ったとしても分子としてのデータはありません。
バグ?
実はこのデータファイルにはMassesがありません。
公式のドキュメントには masses=True のオプションがあり、おそらくこれをやるとMassesが書かれるのですが上手く動きませんでした。
mdcell.write("methanol_64.data", format='lammps-data', masses=True, units="real", atom_style="full")元素はC H Oなので、仕方ないのでデータファイルの zlo zhi と Atoms の間に手書きします。
Masses
1 12.011
2 1.008
3 15.999そしてこちらのファイルをReaxFFで計算してみました。
コメント