PythonのASE (Atomic Simulation Environment) でQuantum ESPRESSOのデータファイルを作って、pw.xによる第一原理MD計算をやってみます。
cp.xのほうの計算はこちら

モデル作成
メタン分子の第一原理MDをやってみます。
モデル作成は以下のコードで行いました。
from ase import Atoms
from ase.visualize import view
from ase.build import molecule
from ase.units import mol
import numpy as np
CH4 = molecule("CH4")
unitcell_size = 10.0
unitcell = Atoms(symbols=CH4.get_chemical_symbols(), positions=CH4.get_positions(),
cell=np.eye(3) * unitcell_size, pbc=True)
shift = [0.5*unitcell_size]*3
unitcell.set_positions(unitcell.get_positions() + shift)
mdcell = unitcell * [1, 1, 1]
density = (np.sum(mdcell.get_masses())/mol) / (mdcell.get_volume()*1e-24)
print(density, "[g/cm3]")
mdcell.write("methane1.in", format='espresso-in')
view(mdcell)これでメタン1分子のセルが作られ、methane1.inファイルが作られます。
unitcell_sizeでセルの1辺の長さをÅ単位で指定しています。密度をprintしており、今回の場合は 0.026 g/cm3でした。
工夫としては、shiftのところでセルサイズの0.5倍分の移動をすることで分子をセルの中央に置いています。(これがないとセルの頂点に1分子置かれる形になります)
また最後のviewで可視化されます。
なお、mdcell = unitcell * [1, 1, 1]の1, 1, 1を2, 2, 2にするとこのセルを2x2x2倍して分子数が8個になります。
入力ファイル編集
作成されたmethane1.inに、以下の内容になるようにいくつかキーワードを追記します。
- NVEアンサンブル(初期温度300K)
- 時間刻み幅 約1fs
- 総ステップ数 50
赤字が追記部分です。
&CONTROL
calculation='md'
dt=20
nstep=50
/
&SYSTEM
ntyp = 2
nat = 5
ibrav = 0
nosym=.true.
/
&ELECTRONS
/
&IONS
ion_temperature='initial'
tempw=300.0
/
&CELL
/
ATOMIC_SPECIES
C 12.011 C.pbe-n-kjpaw_psl.1.0.0.UPF
H 1.008 H.pbe-rrkjus_psl.1.0.0.UPF
K_POINTS gamma
CELL_PARAMETERS angstrom
10.00000000000000 0.00000000000000 0.00000000000000
0.00000000000000 10.00000000000000 0.00000000000000
0.00000000000000 0.00000000000000 10.00000000000000
ATOMIC_POSITIONS angstrom
C 5.0000000000 5.0000000000 5.0000000000
H 5.6291180000 5.6291180000 5.6291180000
H 4.3708820000 4.3708820000 5.6291180000
H 5.6291180000 4.3708820000 4.3708820000
H 4.3708820000 5.6291180000 4.3708820000
時間刻み幅dtは 20×0.048378 [fs]、つまり0.9675 fsです。(約1fs)
デフォルトが20なのでこの通りにしていますが、重い元素のMDを想定したものだと思われます。
今回のように水素があるような計算ではもっと小さい値のほうがいいと思います。
また、擬ポテンシャルファイル置き場は $HOME/espresso/pseudo か$ESPRESSO_PSEUDOで指定していればこのままで動きますが、そうでない場合は
&CONTROL
calculation='md'
dt=20
nstep=50
pseudo_dir='./'
/のようにしてカレントディレクトリなどにファイルを置くといいと思います。
実行結果
以下のコマンドで実行して2分18秒くらいで終わりました。
$ mpiexec -n 8 pw.x -in methane1.in | tee methane1.out結果をASEで可視化します。
$ ase guiでGUIを起動し、FileメニューOpenを開き、outファイルをQuantum espresso out形式で開きます。
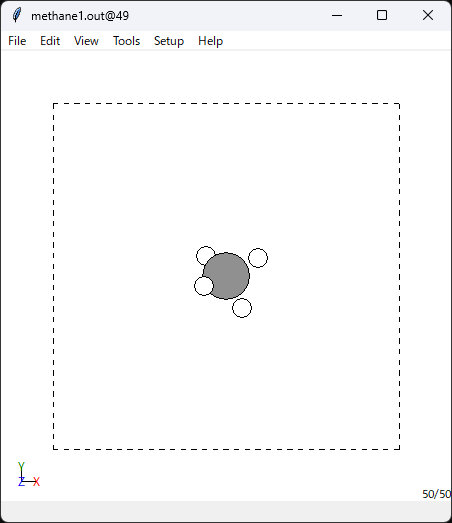
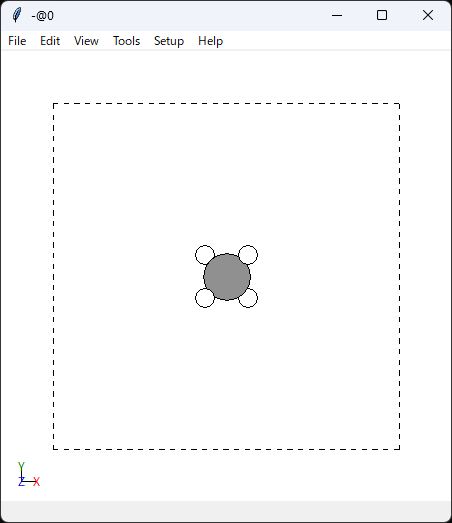
右下に50/50と出ていますので、nstep=50の50フレームが読み込まれています。
そして動画の通り、ちゃんと分子が動いています。
ちなみに温度制御はしていませんが、
$ cat methane1.out |grep temperature
Starting temperature = 300.00 K
temperature is set once at start
temperature = 300.00000000 K
temperature = 266.57370035 K
temperature = 171.87885147 K
temperature = 96.03177706 K
temperature = 86.97776782 K
:
:
temperature = 193.77107145 K
temperature = 166.19831022 K
temperature = 141.05871910 K
temperature = 126.88407794 K
temperature = 122.06809200 Kのように温度が揺らいでいます。まあ分子数が1個なので温度なんてどうでもいいですね。
何はともあれASEで作ったモデルの第一原理MDをQEで計算してASEで可視化できました。
コメント