
PythonのASE (Atomic Simulation Environment) でQuantum ESPRESSOのデータファイルを作って、cp.xによる第一原理MD計算をやってみます。
入力データ作成と計算実行
以下の流れで進みます。
- ASEで入力構造を作成し、第一段階と第二段階の入力ファイルを作成。
- 第一段階の計算(原子を動かさずに電子状態のみを計算)
- 第二段階の計算(原子を動かすMD計算)
詳しい原理や計算キーワードについては他の詳しい記事などを参考にしてください。本記事では、とりあえず動けばいいというレベルで解説します。
ASEで入力構造を作成する部分については以下の記事がベースになっています。
そして第一段階の入力ファイルは以下です。
&CONTROL
calculation='cp'
restart_mode='from_scratch'
dt=5.0
nstep=50
pseudo_dir='./'
/
&SYSTEM
ntyp = 2
nat = 5
ibrav = 0
nosym=.true.
nr1b=16
nr2b=16
nr3b=16
ecutwfc=20.0
/
&ELECTRONS
electron_dynamics='damp'
electron_damping=0.2
/
&IONS
ion_dynamics='none'
/
&CELL
/
ATOMIC_SPECIES
C 12.011 C.pbe-n-rrkjus_psl.1.0.0.UPF
H 1.008 H.pbe-rrkjus_psl.1.0.0.UPF
CELL_PARAMETERS angstrom
10.00000000000000 0.00000000000000 0.00000000000000
0.00000000000000 10.00000000000000 0.00000000000000
0.00000000000000 0.00000000000000 10.00000000000000
ATOMIC_POSITIONS angstrom
C 5.0000000000 5.0000000000 5.0000000000
H 5.6291180000 5.6291180000 5.6291180000
H 4.3708820000 4.3708820000 5.6291180000
H 5.6291180000 4.3708820000 4.3708820000
H 4.3708820000 5.6291180000 4.3708820000
そして第二段階のファイルでは以下の部分を書き換えます。
- &CONTROL
- restart_modeを’from_scratch’から’reset_counters’にする。(第一段階の計算ステップ数カウンターをリセットして、第一段階の計算の続きから計算する)
- nstepにMD計算ステップ数を設定する。
- &ELECTRONS
- electron_dynamicsを’verlet’にする。(ほかの設定もあるが詳しくは知らない)
- &IONS
- ion_dynamicsを’none’から’verlet’にする。(第一段階では原子を動かさなかったが第二段階ではVerlet法で動かす)
- ion_temperatureを’nose’にする。(温度制御を能勢の方法で行う)
- tempwに任意の温度を設定する。(デフォルトでは300)
それ以外は第一段階と同じだと思いますが、エラーが出る場合やより計算内容を厳密に設定する場合があると思いますので、公式のドキュメントやexampleのファイルなどを参考にして書き換えてください。
第一段階の続きから計算するわけですが、計算フォルダ内にcp_50.saveのようや名前のフォルダができると思います。続きから計算するためのデータはここに入っています。
またcp.posなどのファイルは、第二段階の結果が第一段階の結果に追記される形になります。第一段階の結果が不要なのであれば、第二段階の計算前にcp.*を削除するといいと思います。
あとはこれらのファイルをcp.x -in input.in のような感じで実行します。
出力データの可視化
ASEで可視化をしたいと思いますが、ASEはQEのcp.xの出力データには対応していません。
ですがASEではQEのinputファイルには対応しているので、初期構造をinputファイルから読み込み、それ以降のトラジェクトリをcp.xのposファイルとcelファイルから作るコードを書きました。
from ase import Atoms
from ase.io import read, write
from ase.io.trajectory import Trajectory
from ase.units import Bohr
#input
qe_input = "input_2.in"
cel_file = "cp.cel"
pos_file = "cp.pos"
#output
traj_file = "ase.traj"
input_atoms = read(qe_input, format="espresso-in")
symbols = input_atoms.get_chemical_symbols()
Natoms = len(symbols)
def read_file(filename, num_data):
f = open(filename, "r")
lines = f.readlines()
f.close()
data1 = []
data2 = []
data2tmp = []
data2_count = 0
for line in lines:
line = line.replace("\n", "")
values = line.split()
if len(values) == 2:
data1.append(values)
if len(values) ==3:
data2tmp.append(values)
data2_count += 1
if data2_count == num_data:
data2.append(data2tmp)
data2_count = 0
data2tmp = []
return data1, data2
step_time, cell_H = read_file(cel_file, 3)
cel = []
for H in cell_H:
a = float(H[0][0]) * Bohr
b = float(H[1][1]) * Bohr
c = float(H[2][2]) * Bohr
cel.append([a, b, c])
step_time, positions = read_file(pos_file, Natoms)
traj = Trajectory(traj_file, "w")
traj.write(atoms=input_atoms)
for i, pos in enumerate(positions):
xyz = []
for j in range(Natoms):
x, y, z = pos[j]
x = float(x) * Bohr
y = float(y) * Bohr
z = float(z) * Bohr
xyz.append([x, y, z])
snapshot = Atoms(symbols, xyz)
snapshot.set_cell(cel[i])
snapshot.set_pbc(True)
traj.write(atoms=snapshot)
traj.close()
難しいことはやっていません。posやcelファイルはどちらも「ステップ数」と「データの中身」を繰り返し記録している構造なので、それを読み込む関数をdefしています。
posファイルとcelファイルには初期構造が記録されていないので、最初のトラジェクトリは入力ファイルのものを使用しています。
あとは最後のforループでASEのトラジェクトリを作って、ファイルをcloseします。
注意点として、長さの単位がposファイルとcelファイルではボーアになっているので、それをÅに直す定数を掛け算しています。
これを実行して最終的に出力されるase.trajファイルを以下のコマンドで読むことができます。
ase gui ase.traj結果はこんな感じでした。
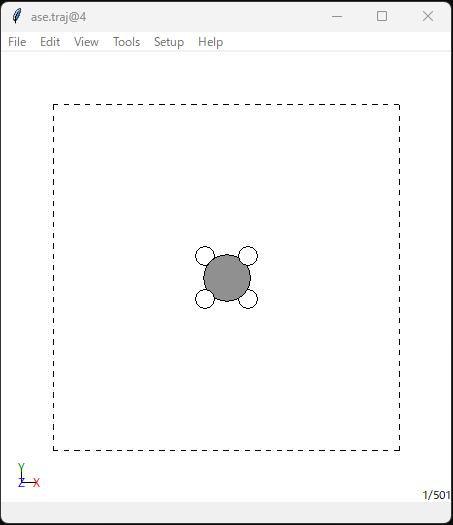
それっぽい動きをしていますね。またASEでの描画も問題なさそうです。
コメント
第一段階と第二段階の入力ファイルは、同じフォルダに作成すればいいでしょうか?
その後、第一段階の計算を実施し、第二段階の計算を実施とすればいいですか?
2つの入力ファイルは同じフォルダに置いて計算を実行します。
第一段階の計算が終わるとcp_50.saveのような名前のフォルダとcp.posなどのファイルができます。
フォルダのほうは第二段階で使用するデータが入っているので、同じフォルダで実行するのが無難です。フォルダの指定は入力ファイル内で指定することもできるはずです。
ファイルのほうは、第一段階のデータに追記する形で第二段階の結果が記録されます。第一段階のデータが不要な場合は、第二段階が始まる前に削除しておくといいと思います。
ご教授頂きありがとうございました。
おかげで計算できるようになりました。
現在、Tesla P100で遊んでいます。
冷却が不充分なのか、今のところ、4070TiのほうがP100より速い結果です。
倍精度計算(fp64)で真価を発揮できるといいのですが、冷却が思ったより大変です。
現在、フル稼働時は79℃です。HP見る限り、70℃くらいに冷却しないと4.7Tflops出ないらしいです。(80℃では500Mflopsくらいになるらしい)
VRAM16GBくらいしか今のところ恩恵がありません。。。
スパコン向けGPUと言えど最新モデルには敵わないんですね。冷却も夏は厳しそう…
LLMを動かす場合だとVRAMが必要ですが、4070Tiもそこそこありますし、活用が難しいですね。
おはようございます。
P100は、ebayで新品が4万円(送料込み)と格安でした。
8年前の発売当初は、100万円だったようです。
Quantum Espressoで比較すると、
4070Ti:1時間
P100:1時間30分
くらいな感じでした。
メモリー多いほうが大きなモデル計算できますし、2つあっても仕方ないので、高く売れるうちに4070Tiは5080の軍資金にするため、売ってしまいました。
これ以上円高になると新品価格が下がって、買取価格下がりそうです。