PythonのASE (Atomic Simulation Environment) でLAMMPSのファイルを読み込むと、正しい元素が反映されないことがあります。
LAMMPSは汎用なソルバーであり原子以外のものも計算ができますが、それゆえ原子を原子として認識せず、ある単位である質量が設定された粒だという情報しかファイルには記されていません。
そういうわけでASEがLAMMPSデータの元素を認識できない場合があるので、それを認識させる方法を紹介します。
問題点
こちらの記事のメタノールのデータを使用します。

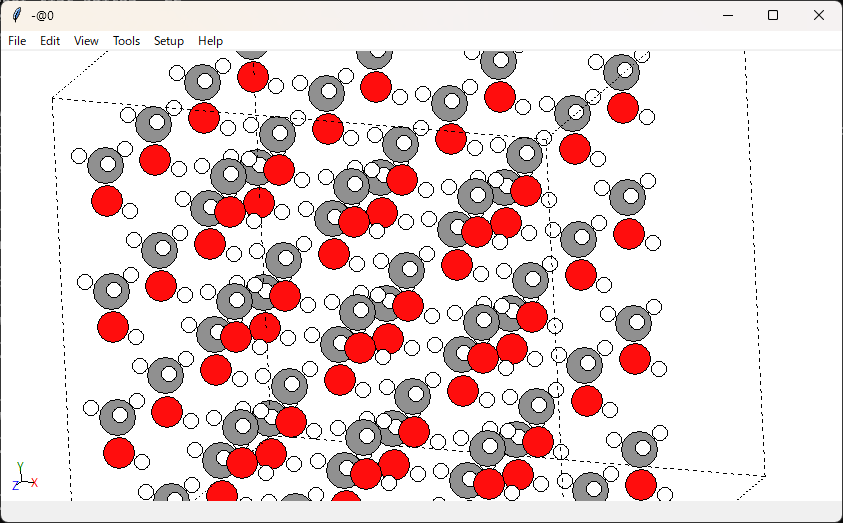
このデータを作った時点では元素は正しく設定され、このように可視化ができます。
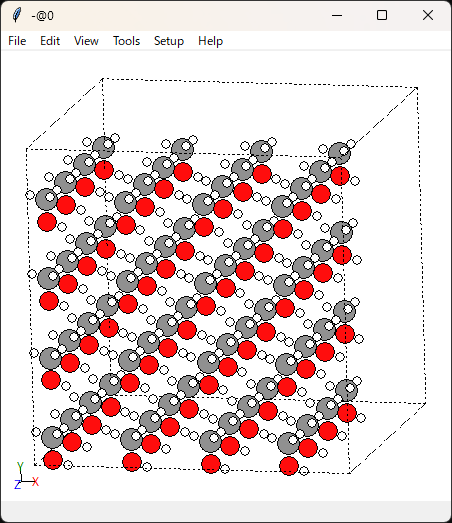
ではこのデータをファイルとして保存し、そのファイルを読み込んでみます。
このように色が変わっていますね。さらにコマンドラインで確認します。
>>> from ase.io import read
>>> atoms = read("methanol_64.data", format="lammps-data")
>>> print(atoms)
Atoms(symbols='H64He256Li64', pbc=True, cell=[20.0, 20.0, 20.0], id=..., initial_charges=..., mmcharges=..., mol-id=..., type=...)メタノールなのにヘリウムとかリチウムが入ってます。
こうなる理由は、ASEがLAMMPSデータの元素を把握していないからです。
データ内の 1番目の元素 2番目の元素 3番目の元素 を 水素 ヘリウム リチウム と原子番号順に読み込んでしまっています。
元素を設定し直す
それでは元素を設定し直します。
実は上記のPythonコードでreadしたデータには質量情報が含まれています。
そのため、データ内の質量と実際の元素の質量をマッチングさせれば正しい元素にすることができます。
from ase.io import read
from ase.data import chemical_symbols, atomic_masses
from ase.visualize import view
import numpy as np
atoms = read("methanol_64.data", format="lammps-data")
correct_symbols = []
for mass in atoms.get_masses():
diff_mass = np.abs(atomic_masses - mass)
atomic_number = np.argmin(diff_mass)
correct_symbols.append(chemical_symbols[atomic_number])
atoms.set_chemical_symbols(correct_symbols)
print(atoms)
view(atoms)2行目でインポートしているchemical_symbolsは全元素の元素記号のリスト、atomic_massesは全元素の質量のリストです。
読み込んだatomsデータの質量でfor文を回して、全元素との差が最も小さいもののインデックス番号(原子番号)を取得し、元素記号リストに入れて元素記号を決定、リストに格納、という流れです。
printの結果は
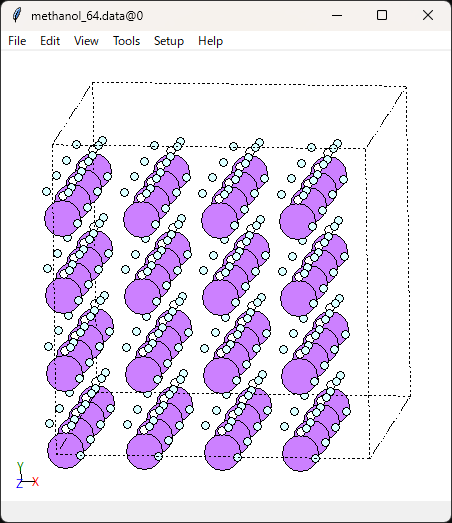
Atoms(symbols='C64H256O64', pbc=True, cell=[20.0, 20.0, 20.0], id=..., initial_charges=..., masses=..., mmcharges=..., mol-id=..., type=...)viewの結果は
ということで、正しい元素に修正することができました。
修正できていない場合は、atoms.get_masses()を実行して質量が正しく読み込まれているかどうかを確認しましょう。
トラジェクトリの場合
トラジェクトリの場合はdumpファイルに質量情報が含まれませんので、先ほどのPythonコードの要領でlammps-dataファイルから元素情報を取得し、トラジェクトリの1枚1枚のスナップショットに元素情報を反映していきます。
from ase.io import read, write
from ase.data import chemical_symbols, atomic_masses
from ase.io.trajectory import Trajectory
from ase.visualize import view
import numpy as np
atoms = read("methanol_64.data", format="lammps-data")
print(atoms.get_chemical_symbols())
correct_symbols = []
for mass in atoms.get_masses():
diff_mass = np.abs(atomic_masses - mass)
atomic_number = np.argmin(diff_mass)
correct_symbols.append(chemical_symbols[atomic_number])
traj_input = read("dump.lammpstrj", format="lammps-dump-text", index=":")
traj_output = Trajectory("ase.traj", "w")
for snapshot in traj_input:
snapshot.set_chemical_symbols(correct_symbols)
traj_output.write(snapshot)
traj_output.close()出力ファイルはase.trajというファイルにしました。
コマンドラインで ase gui ase.traj を実行すると、
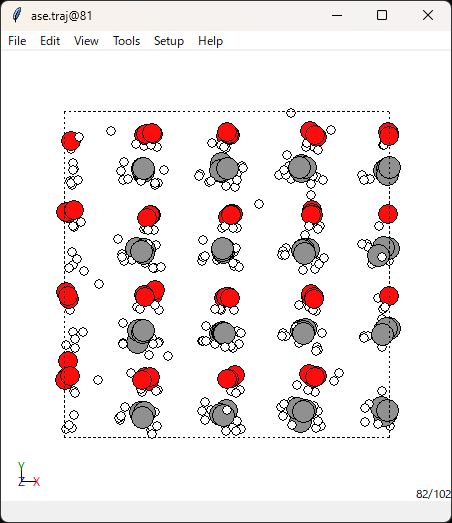
このように、トラジェクトリを正しい元素情報で表示させられます。
コメント