古くからよく使われている剛体水分子モデルであるSPC/Eを使って水の密度を計算してみたいと思います。
SPC/E水分子モデルについて
水が持つ様々な性質を精度よく再現するために、様々な種類の水モデルが考案されています。
その一つがSPC/E (extended simple point charge model, 拡張単純点電荷モデル)です。
元の論文は
Berendsen, Herman JC, J. Raul Grigera, and Tjerk P. Straatsma. “The missing term in effective pair potentials.” Journal of Physical Chemistry 91.24 (1987): 6269-6271.
他の3サイト剛体水分子モデルなども含め、パラメータ等はWikipediaに詳しく載っています。
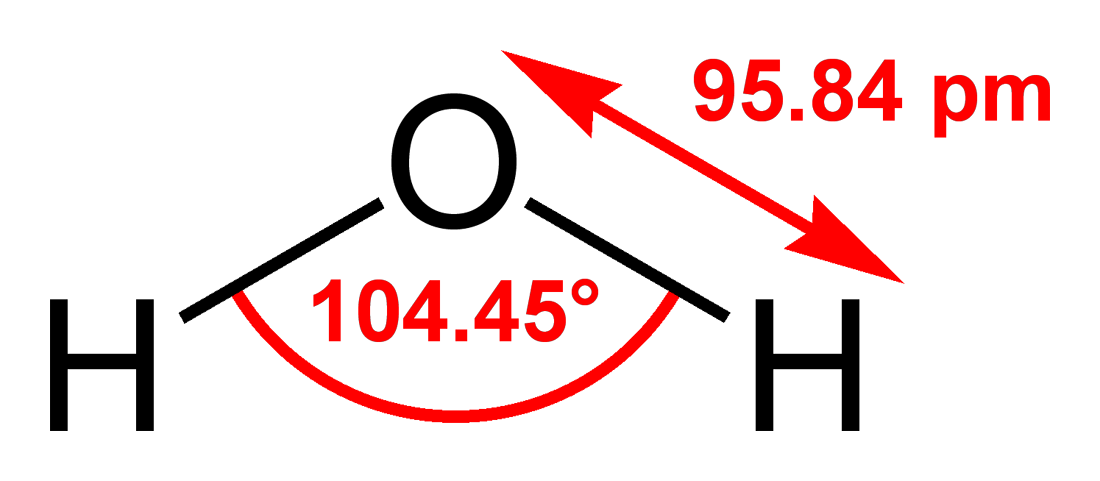
計算内容
LAMMPS公式ドキュメントのサンプルコード
LAMMPSの公式ドキュメントページにSPC/Eを使った計算の例が載っています。

今回はこちらのページの内容をベースにして計算していきます。
ファイルの準備
spce.molファイルを用意します。
# Water molecule. SPC/E geometry
3 atoms
2 bonds
1 angles
Coords
1 0.00000 -0.06461 0.00000
2 0.81649 0.51275 0.00000
3 -0.81649 0.51275 0.00000
Types
1 1 # O
2 2 # H
3 2 # H
Charges
1 -0.8476
2 0.4238
3 0.4238
Bonds
1 1 1 2
2 1 1 3
Angles
1 1 2 1 3
Shake Flags
1 1
2 1
3 1
Shake Atoms
1 1 2 3
2 1 2 3
3 1 2 3
Shake Bond Types
1 1 1 1
2 1 1 1
3 1 1 1
Special Bond Counts
1 2 0 0
2 1 1 0
3 1 1 0
Special Bonds
1 2 3
2 1 3
3 1 2このspce.molは、計算入力ファイルから呼び出す形になります。
今回は水分子を500個用意しようと思います。
密度を1.0g/cm3とすると、水分子1個当たりの体積は29.89Å3です。
500倍した場合、立方体セルは一辺24.6Åくらいなので、少し余裕を持たせた適当な大きさ(-12.7 – 12.7)で作ります。
また、温度はコマンドライン引数で指定できるように変数${temperature}としています。
これは初速度を与えるvelocity allの行と、計算中の温度制御のfix npt行に書きました。
pair_styleはEwald法を使いたいため、lj/cut/coul/cut からlj/cut/coul/long に変更し、kspace_styleを指定しています。
Minimizeは省略、shakeのログも出力しない設定で、以下のようなファイルになりました。
units real
atom_style full
region box block -12.7 12.7 -12.7 12.7 -12.7 12.7
create_box 2 box bond/types 1 angle/types 1 &
extra/bond/per/atom 2 extra/angle/per/atom 1 extra/special/per/atom 2
mass 1 15.9994
mass 2 1.008
pair_style lj/cut/coul/long 10.0 10.0
pair_coeff 1 1 0.1553 3.166
pair_coeff 1 2 0.0 1.0
pair_coeff 2 2 0.0 1.0
kspace_style ewald 1.0e-4
bond_style zero
bond_coeff 1 1.0
angle_style zero
angle_coeff 1 109.47
molecule water spce.mol
create_atoms 0 random 500 34564 NULL mol water 25367 overlap 1.33
timestep 1.0
fix rigid all shake 0.0001 20 0 b 1 a 1
velocity all create ${temperature} 5463576
fix integrate all npt temp ${temperature} ${temperature} 100.0 iso 1.0 1.0 1000.0
thermo_style custom step temp press etotal density pe ke
thermo 1000
run 100000 upto
write_data spce.data nocoeff
ファイル名をin.spceとして、準備完了です。
追記
上の例は原子の定義が1:酸素、2:水素の場合ですが、1:水素、2:酸素の場合はpair_coeffのセクションが以下のようになります。
pair_style lj/cut/coul/long 10.0 10.0
pair_coeff 1 1 0.0 1.0
pair_coeff 1 2 0.0 1.0
pair_coeff 2 2 0.1553 3.166計算
以下のコマンドを実行します。
なお-varオプションにより、計算ファイル内の ${temperature} に 298.15 が代入されます。
このコマンドラインの数値を書き換えるだけで温度設定を変更できます。
バッチファイルやシェルスクリプトで温度を連番で指定して計算ができるので便利です。
lmp -in in.spce -var temperature 298.15出力の一部を以下に貼っています。
molecule water spce.mol
Read molecule template water:
1 molecules
0 fragments
3 atoms with max type 2
2 bonds with max type 1
1 angles with max type 1
0 dihedrals with max type 0
0 impropers with max type 0
create_atoms 0 random 500 34564 NULL mol water 25367 overlap 1.33
Created 1500 atoms
using lattice units in orthogonal box = (-12.7 -12.7 -12.7) to (12.7 12.7 12.7)
create_atoms CPU = 0.030 secondsこちらは分子をランダム生成する部分です。
1500個の原子(500個の水分子)が生成されていることが確認できます。
セルが小さいとoverlapの設定により分子が削除されることがあり、その場合は分子数が減ります。
Step Temp Press TotEng Density PotEng KinEng
0 298.15 127252.61 8295.0578 0.91277098 7407.218 887.83979
1000 278.50803 -1816.5944 -2536.7322 0.83690492 -3366.0815 829.34937
2000 297.62152 767.54138 -764.51772 0.85248793 -1650.7838 886.26605
3000 294.41703 -940.33075 -1482.4847 0.85726488 -2359.2084 876.72365
4000 303.02266 -1029.3307 -2280.3291 0.86752698 -3182.6788 902.34974
5000 309.55568 -1625.7826 -2763.2554 0.88416182 -3685.0594 921.80395
:
:
495000 299.14584 1735.5836 -1348.4354 0.98740201 -2239.2406 890.80522
496000 298.58532 3505.1897 -661.91297 0.9898038 -1551.0491 889.1361
497000 303.76877 -61.1881 -2650.2411 0.9864683 -3554.8126 904.57153
498000 303.2069 -508.79768 -2335.7307 0.98587143 -3238.6291 902.89837
499000 300.95014 1437.7024 -1446.994 0.98625999 -2343.1721 896.17812
500000 308.30472 1458.3828 -1400.4053 0.98735969 -2318.4841 918.07881
Loop time of 770.237 on 4 procs for 500000 steps with 1500 atoms
Performance: 56.087 ns/day, 0.428 hours/ns, 649.151 timesteps/s, 973.726 katom-step/s
9.4% CPU use with 4 MPI tasks x 1 OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 454.23 | 463.55 | 473.13 | 35.4 | 60.18
Bond | 0.23932 | 0.24539 | 0.25098 | 0.8 | 0.03
Neigh | 0.020919 | 0.021483 | 0.022327 | 0.4 | 0.00
Comm | 55.791 | 55.9 | 56.019 | 1.1 | 7.26
Output | 0.014818 | 0.015891 | 0.018985 | 1.4 | 0.00
Modify | 232.24 | 241.71 | 251 | 48.8 | 31.38
Other | | 8.79 | | | 1.14出力項目はthermo_style custom step temp press etotal density pe keにある通り、ステップ、温度、圧力、全エネルギー、密度、ポテンシャルエネルギー、運動エネルギーです。
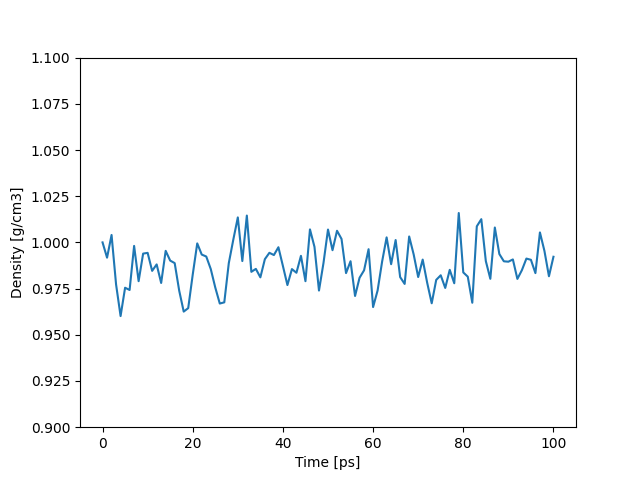
密度をプロットしたところ以下のようになりました。初期密度からほぼ変わらず1近傍をキープしています。
計算後半部分の平均密度は0.9888 g/cm3でした。
データはPythonで処理しました。
使用したコードはこちらの記事で紹介しています。
まとめ
LAMMPS公式で紹介されているSPC/E水モデルの計算ファイルに手を加えて、密度の計算をやってみました。
水専用に作られたパラメータなので密度が合うのは当然ですが、簡単に再現できました。
コメント