LAMMPSで反応力場 ReaxFFを使ってみます。
使用するデータ
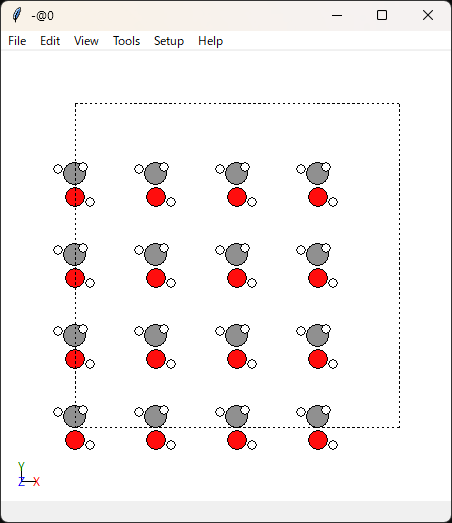
使用するデータはこちらで作ったものです。

このmethanol_64.dataファイルを初期構造とします。
Minimize計算
ReaxFFを指定するのは以下の3行です。
pair_style reaxff NULL checkqeq yes
pair_coeff * * ffield.reax.cho C O H
fix 1 all qeq/reax 1 0.0 10.0 1.0e-6 reaxffここではReaxFFとQEQ(電荷平衡法)を適用しています。
またpair_coeffで指定しているffield.reax.choは、環境変数LAMMPS_POTENTIALSのフォルダかカレントディレクトリに置く必要があります。
以下のmethanol_64_minimize.inファイルは構造最適化(エネルギー最小化)で使用します。
atom_style full
units real
boundary p p p
read_data methanol_64.data
pair_style reaxff NULL checkqeq yes
pair_coeff * * ffield.reax.cho C O H
fix 1 all qeq/reax 1 0.0 10.0 1.0e-6 reaxff
thermo_style custom step cpu vol pe fmax fnorm
min_style cg
min_modify dmax 0.100000 line quadratic
minimize 1.0e-6 1.0e-8 1000 10000
write_data methanol_64_min.dataこのminimize計算では、初期構造の中で不安定な部分(大きい力がかかっている部分)を緩和して、基準以下までエネルギーが下がったところの構造をwrite_dataで書き出してくれます。
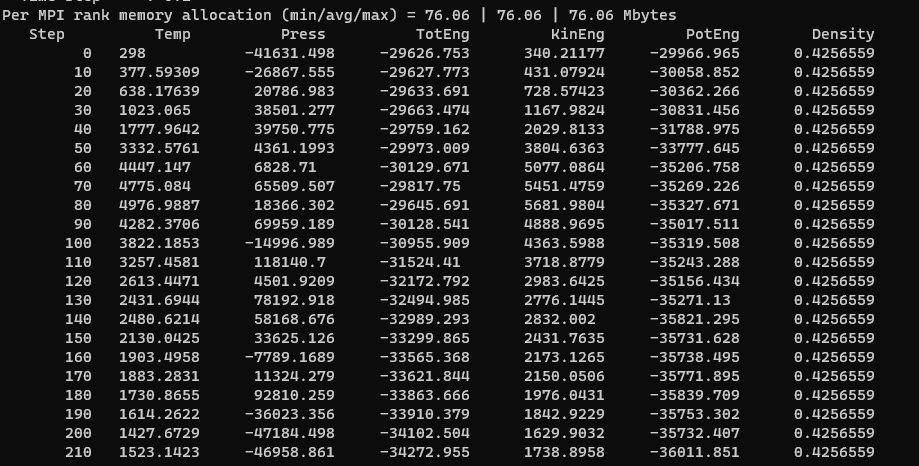
計算の出力は
Step CPU Volume PotEng Fmax Fnorm
0 0 8000 25803.493 1396.3662 20939.984
1 0.043466 8000 -7614.5601 877.92025 11804.323
2 0.088331 8000 -20367.211 705.45629 9066.0746
3 0.133091 8000 -27257.653 363.82267 4580.6323
4 0.216904 8000 -29966.964 121.78475 1506.7081
5 0.721046 8000 -29966.965 121.87887 1508.7675
Loop time of 0.721453 on 1 procs for 5 steps with 384 atomsたったの5ステップで収束しました。
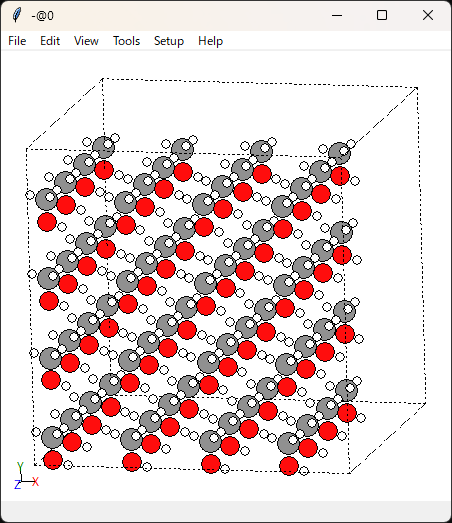
上の画像の通り分子同士が干渉していないので、分子内の構造が少し動いた程度だと思います。
MD計算
Minimize計算の結果の構造はmethanol_64_min.dataに保存されているので、それを初期構造としてNVTアンサンブルのMD計算を少しだけ流してみます。
atom_style full
units real
boundary p p p
read_data methanol_64_min.data
pair_style reaxff NULL checkqeq yes
pair_coeff * * ffield.reax.cho C O H
fix 1 all qeq/reax 1 0.0 10.0 1.0e-6 reaxff
timestep 0.2
run_style verlet
atom_modify sort 0 0.0
velocity all create 298.0 4928459
fix integrate all nvt temp 298.0 298.0 20.0
thermo_style custom step temp press etotal ke pe density
thermo 10
dump myDump all atom 10 dump.lammpstrj
dump_modify myDump sort id
run 1000
write_data methanol_64_1st.data
計算を実行して、最後の1000step目の構造をVMDで可視化してみます。
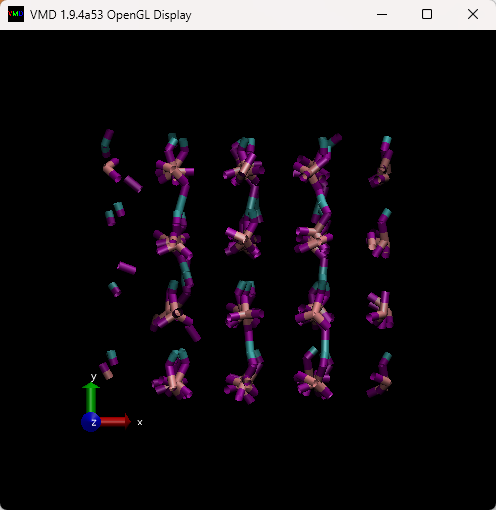
分子が崩壊している様子もないので、たぶんちゃんと計算できています。
ここから長時間の計算を行ってNPTアンサンブルで密度緩和をしていくとよいと思います。
なお計算速度は通常の古典力場のMD計算よりも遅いので覚悟してください。
追記
計算結果をASEで可視化する方法はこちらです。
コメント